Analytical Data
-
基因名
CFTR
- Application
-
别名
CFTR;ABCC7;Cystic fibrosis transmembrane conductance regulator
-
种属
Human
-
表达系统
E. coli
-
标签
His tag N-Terminus
-
纯度
Greater than 90% as determined by SDS-PAGE.
-
蛋白编号
P13569
-
表达区间
1381-1480aa
-
氨基酸序列
YQIIRRTLKQAFADCTVILCEHRIEAMLECQQFLVIEENKVRQYDSIQKL LNERSLFRQAISPSDRVKLFPHRNSSKCKSKPQIAALKEETEEEVQDTRL
-
分子量
37 kDa
-
内毒素
< 1.0 EU per μg protein as determined by the LAL method.
-
性状
Freeze-dried powder
-
缓冲液
PBS, pH7.4, containing 0.01% SKL, 1mM DTT, 5% Trehalose and Proclin300.
-
复溶方法
Reconstitute in ddH2O to a concentration of 0.1-0.5 mg/mL. Do not vortex.
- 个性化定制
-
稳定性测试
The thermal stability is described by the loss rate. The loss rate was determined by accelerated thermal degradation test, that is, incubate the protein at 37℃ for 48h, and no obvious degradation and precipitation were observed. The loss rate isless than 8% within the expiration date under appropriate storage condition.
-
保存条件 & 期限
Samples are stable for up to twelve months from date of receipt at -20℃ to -80℃. Store it under sterile conditions at -20℃ to -80℃. It is recommended that the protein be aliquoted for optimal storage. Avoid repeated freeze-thaw cycles.
-
运输条件
In general, recombinant proteins are supplied as lyophilized powder and shipped at ambient temperature. For bulk packages, the proteins are provided as frozen liquid and shipped with blue ice, unless otherwise requested by the customer.
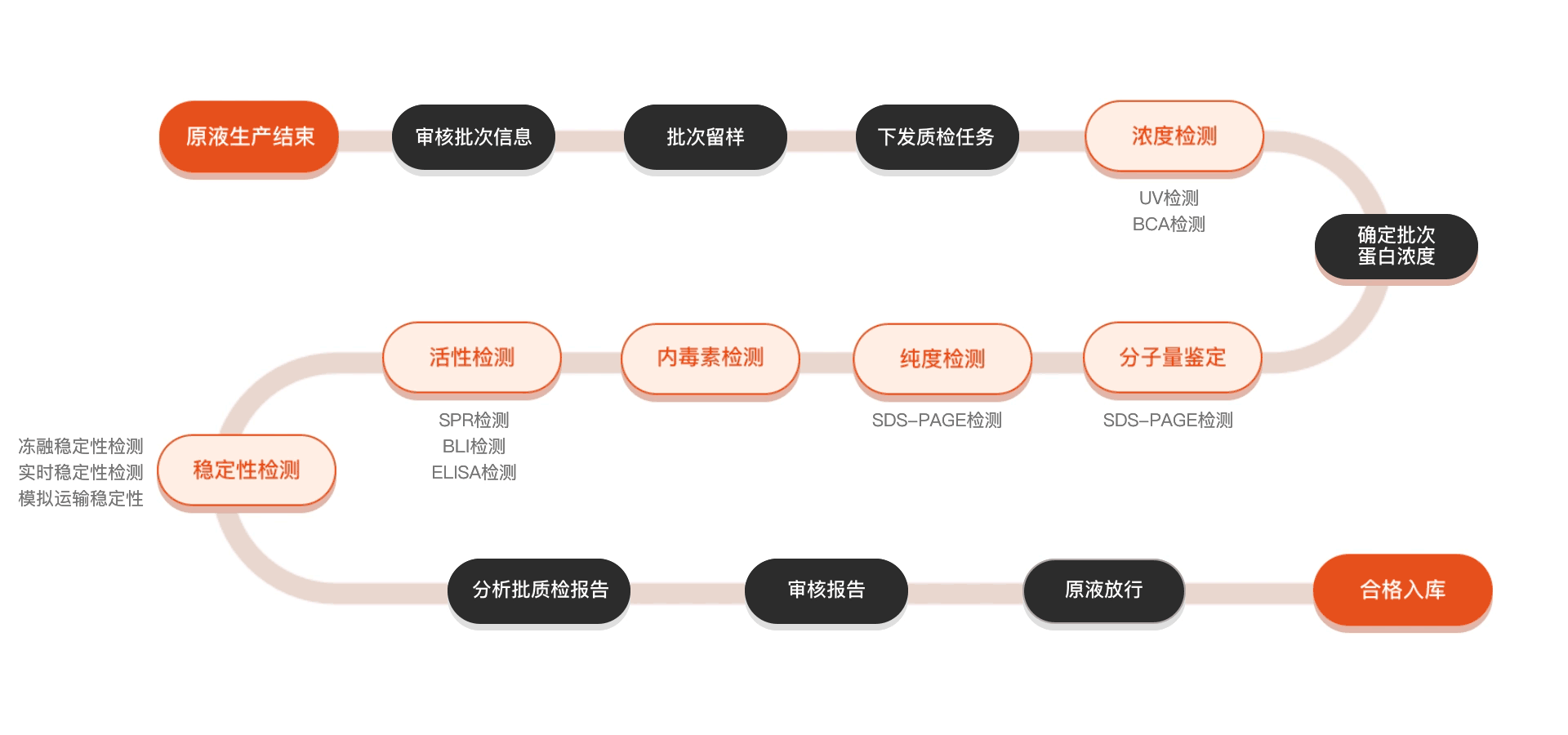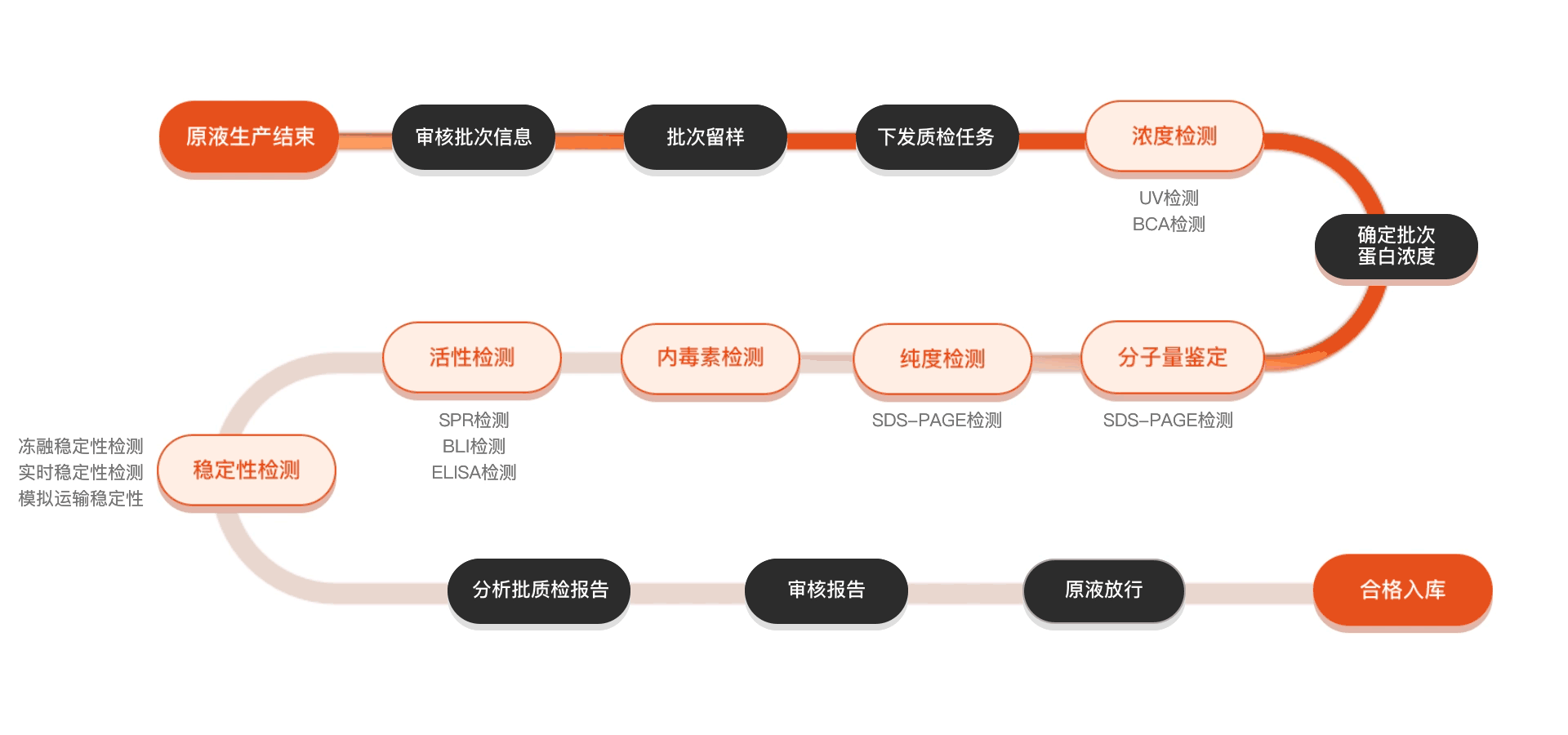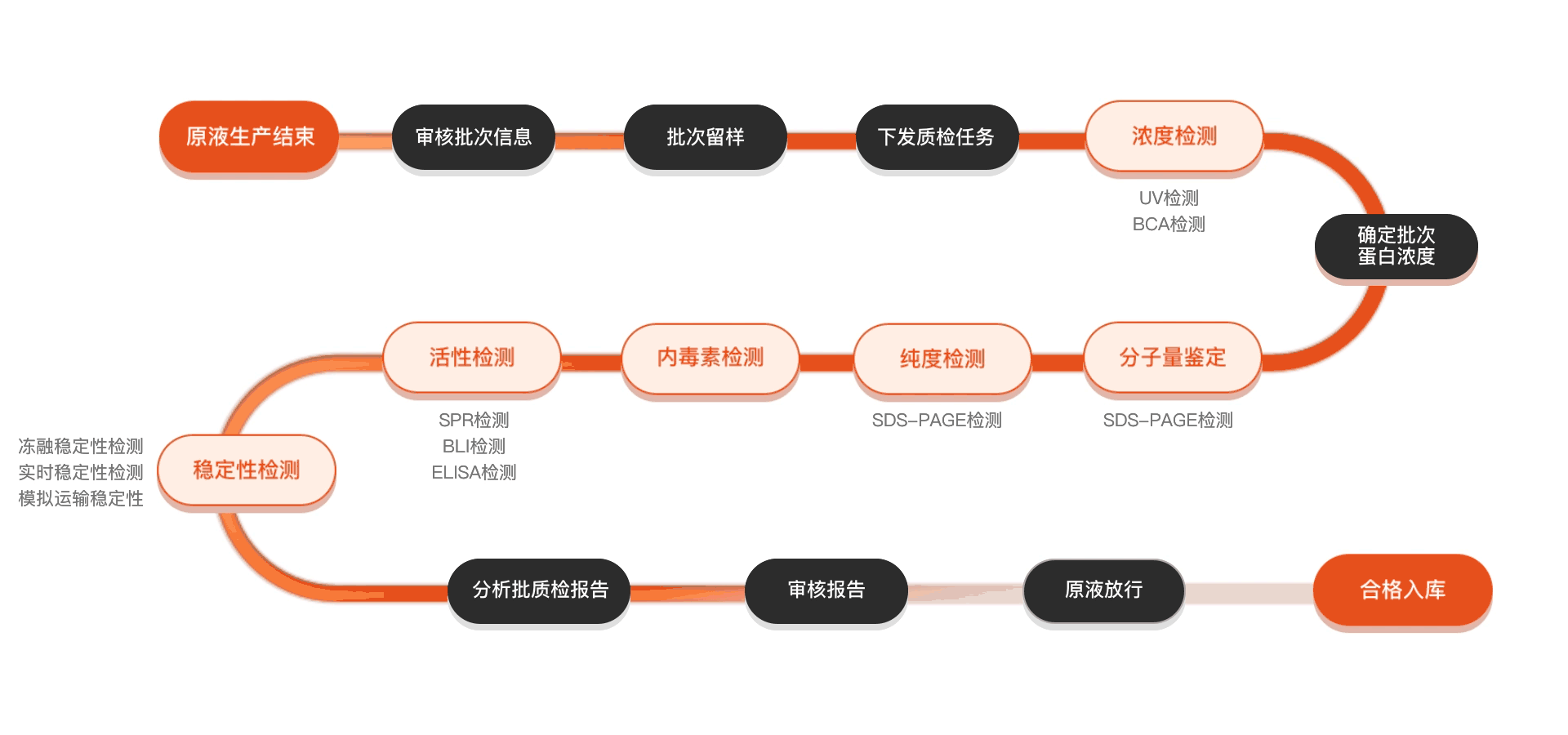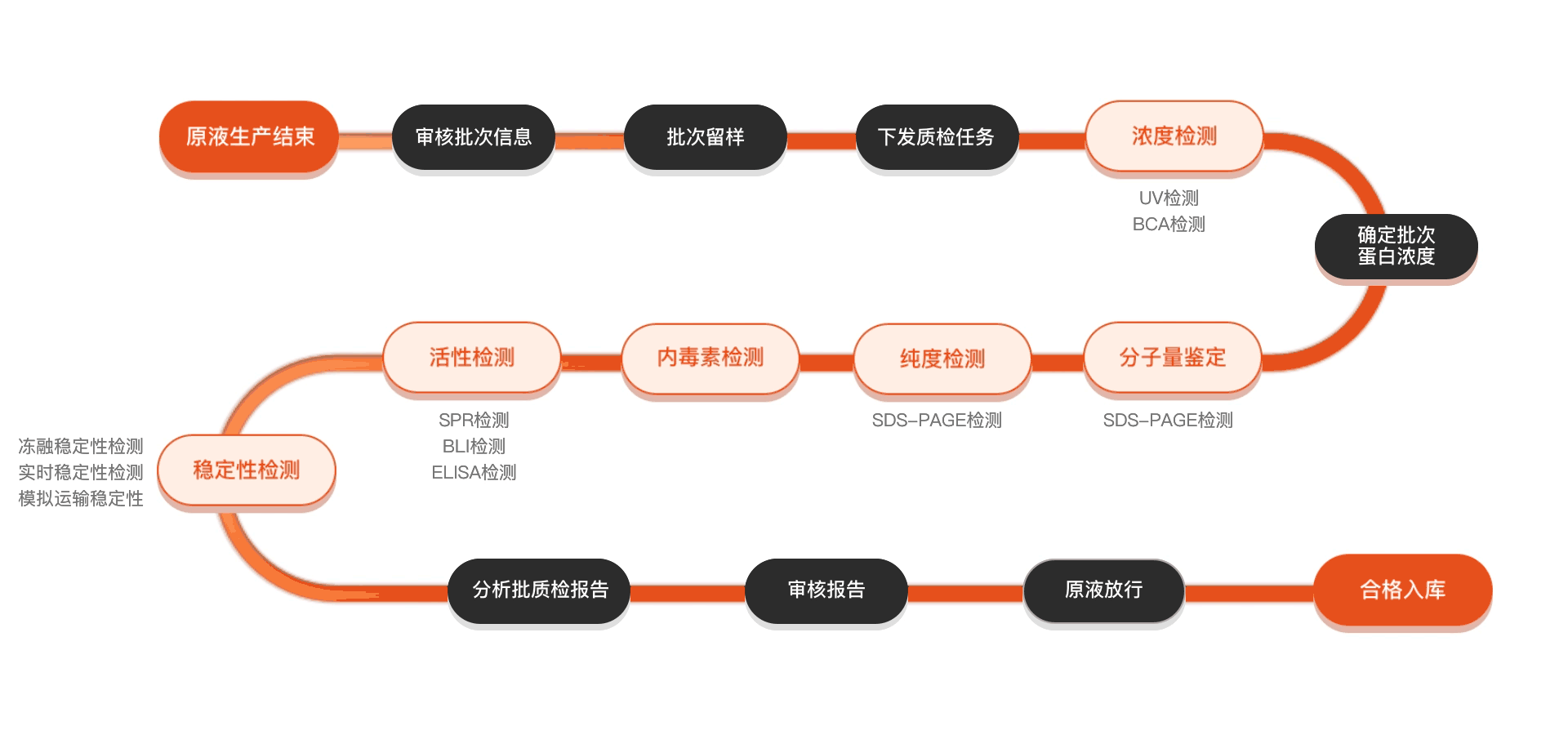
Quality inspection process
Related Products






Protein Description
The cystic fibrosis transmembrane conductance regulator (CFTR) is a vital protein that functions as a chloride channel in epithelial cells, playing a crucial role in maintaining the balance of salt and water across cell membranes. Mutations in the CFTR gene lead to cystic fibrosis (CF), a genetic disorder characterized by thick mucus production, respiratory problems, and impaired pancreatic function. Research on CFTR recombinant proteins has gained significant attention due to their potential to elucidate the mechanisms underlying CFTR function and dysfunction. The study of CFTR's structure, dynamics, and interaction with various cellular components is essential for understanding the molecular basis of CF and developing targeted therapies. By employing techniques such as protein expression in heterologous systems, site-directed mutagenesis, and advanced imaging methods, scientists aim to dissect the channel's gating mechanisms and ion transport properties. Furthermore, the characterization of CFTR recombinant proteins offers insights into how specific mutations affect its function, paving the way for the design of novel pharmacological agents, such as small molecules and gene therapies, that could restore CFTR activity and improve outcomes for individuals with cystic fibrosis. As research progresses, the integration of structural biology, biophysics, and pharmacology continues to enhance our understanding of CFTR, highlighting its significance as a therapeutic target and a model for studying ion channel diseases.